Tumor Classification with Machine Learning
The Challenge: To accurately classify breast cancer tumors as malignant or benign from complex genomic data, providing a potential tool for faster diagnostics.
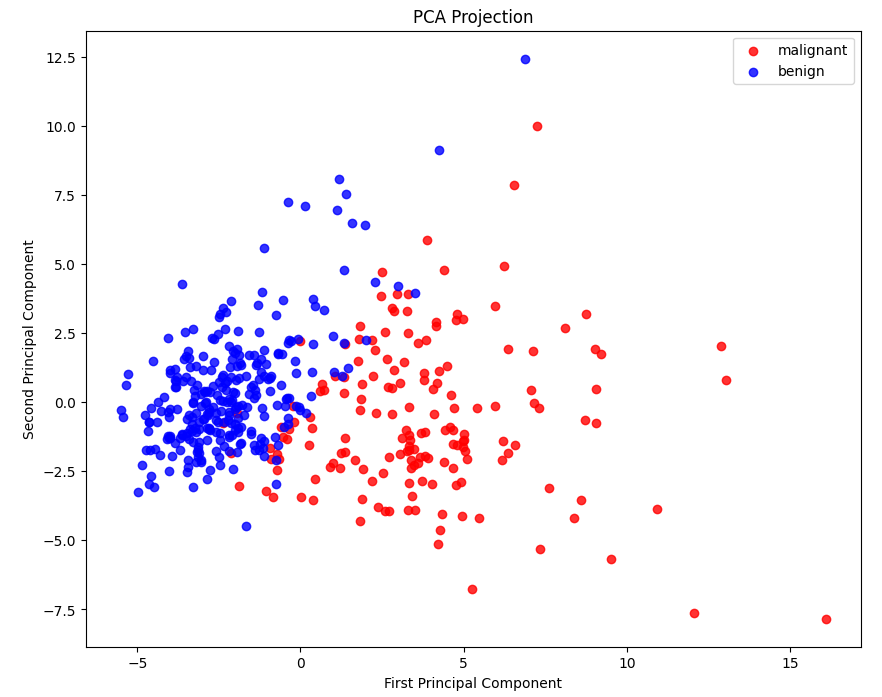
My Approach: I developed an end-to-end pipeline in Python using scikit-learn. This involved rigorous data preprocessing, feature selection using PCA for dimensionality reduction, and training multiple classifiers. An ensemble model (Random Forest and Voting Classifier) was implemented to maximize accuracy and robustness.
The Outcome: The model achieved high classification accuracy. Using Random Forest's feature importance, I was able to identify key genetic markers that were most predictive of tumor malignancy, demonstrating a full cycle of data analysis from raw data to actionable insight.
View Code on GitHub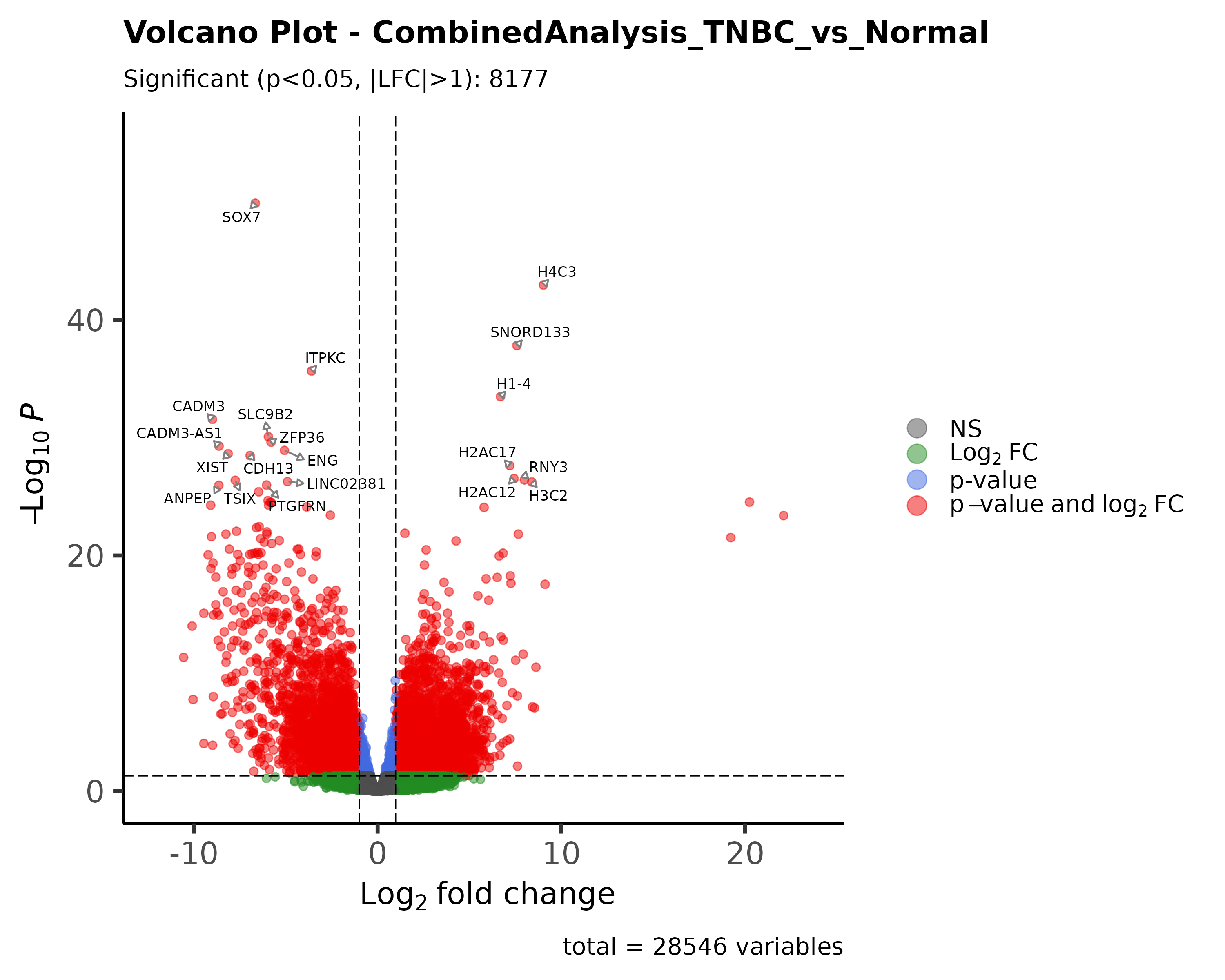
Differential Gene Expression in Breast Cancer
The Challenge: To identify genes that are significantly up- or down-regulated in Triple-Negative Breast Cancer (TNBC) compared to normal tissue, which could reveal potential therapeutic targets.
My Approach: I sourced and integrated three separate datasets from the Gene Expression Omnibus (GEO). Using R and the DESeq2 package, I performed a rigorous differential gene expression analysis pipeline. The results were visualized using advanced plots like volcano plots, heatmaps, and PCA plots for clear interpretation.
The Outcome: The analysis successfully identified a list of statistically significant DEGs. I further explored their functional relationships by constructing a protein-protein interaction (PPI) network using STRINGdb, highlighting key biological pathways implicated in TNBC.
View Code on GitHub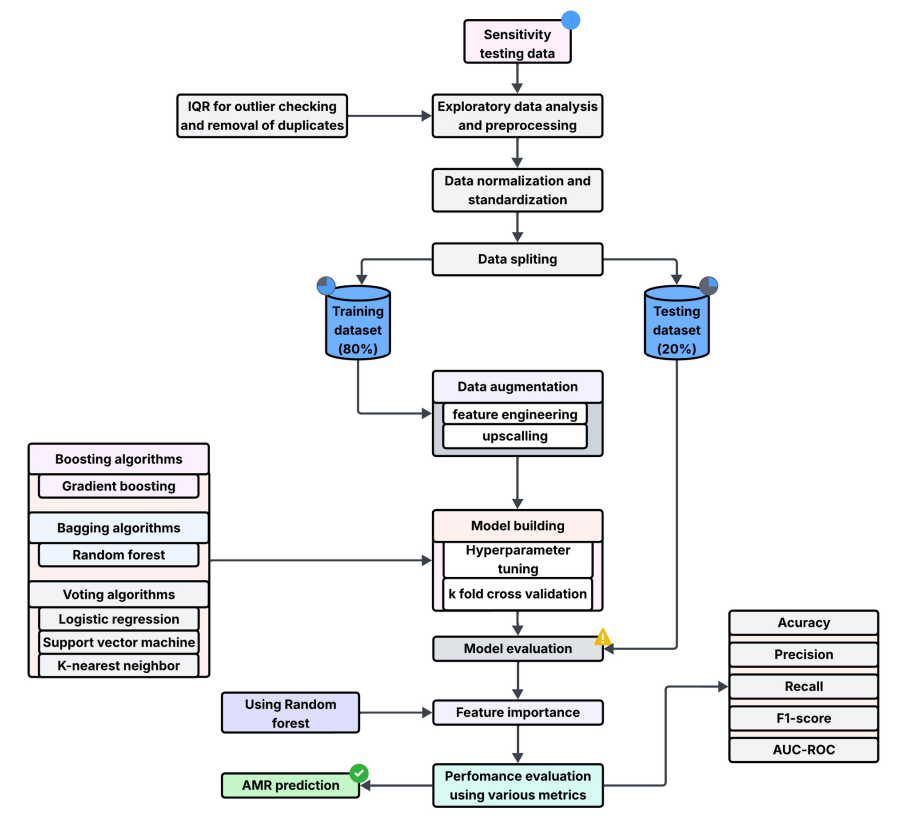
Predictive Model for AMR Surveillance (Thesis)
The Challenge: Antimicrobial Resistance (AMR) is a silent pandemic. The goal of my undergraduate thesis was to develop a proof-of-concept machine learning model to predict AMR patterns from clinical data, aiding in public health surveillance.
My Approach: This project involved collecting and cleaning diverse clinical datasets. I then engineered features relevant to resistance patterns and trained several predictive models to identify the most effective algorithm. The focus was on creating a model that was not only accurate but also interpretable.
The Outcome: The resulting model demonstrated the feasibility of using machine learning for AMR surveillance, laying the groundwork for more advanced, data-driven approaches to combat infectious diseases in the region. The project solidified my skills in the entire machine learning lifecycle, from data collection to model deployment.
Code Not Public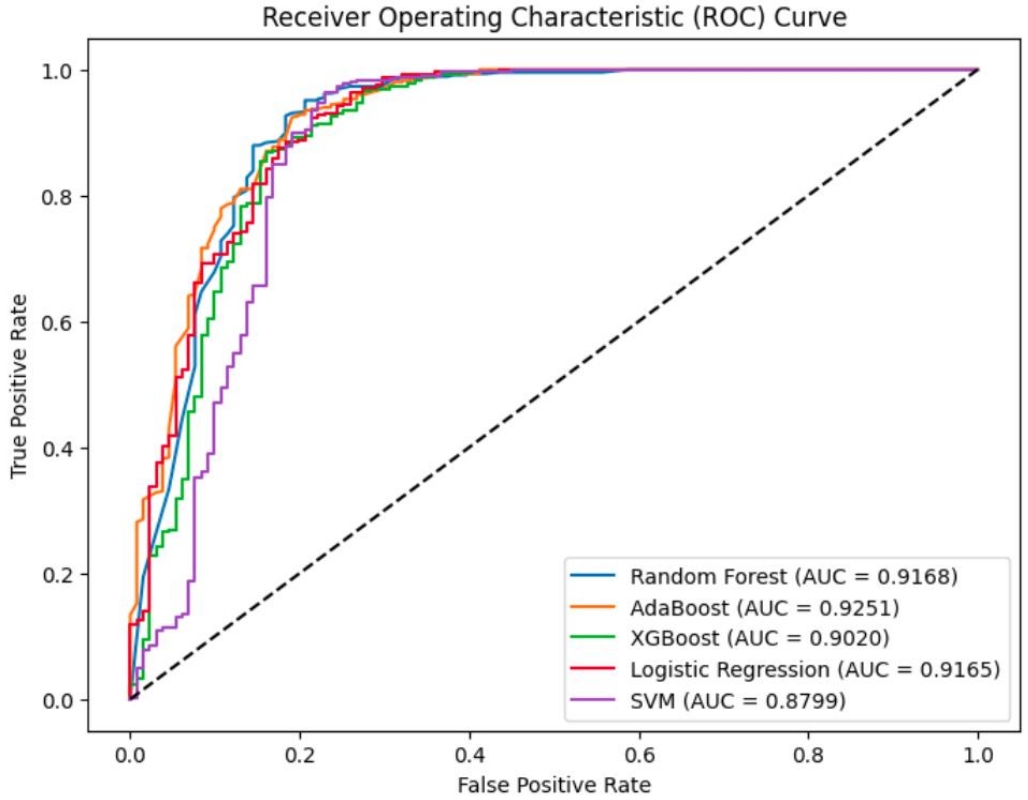
Predicting HIV Viral Load Suppression in Zimbabwe
The Challenge: Despite high treatment coverage in Zimbabwe, a significant portion of HIV-positive adults fail to achieve viral load suppression (VLS). Identifying these at-risk individuals is difficult with traditional methods, and no machine learning models existed specifically for the Zimbabwean context.
My Approach: I leveraged the nationally representative ZIMPHIA 2020 survey dataset of nearly 3,000 patients. The pipeline, built using R and Python, involved rigorous data cleaning, preprocessing, and feature selection using Recursive Feature Elimination (RFE). I developed and conducted a comparative analysis of five distinct machine learning models (including Random Forest, AdaBoost, XGBoost, and SVM) to predict VLS, validated using 10-fold cross-validation.
The Outcome: The Random Forest model delivered the best overall performance, achieving a high AUC of 0.92 and a cross-validated AUC of 0.98. Crucially, the model's feature importance analysis identified CD4 cell count and antiretroviral (ARV) status as the most powerful predictors. This project successfully demonstrates the feasibility of using ML to create a data-driven tool that can help clinicians identify high-risk patients, enabling targeted interventions and optimizing HIV care in resource-limited settings.
View Code on GitHub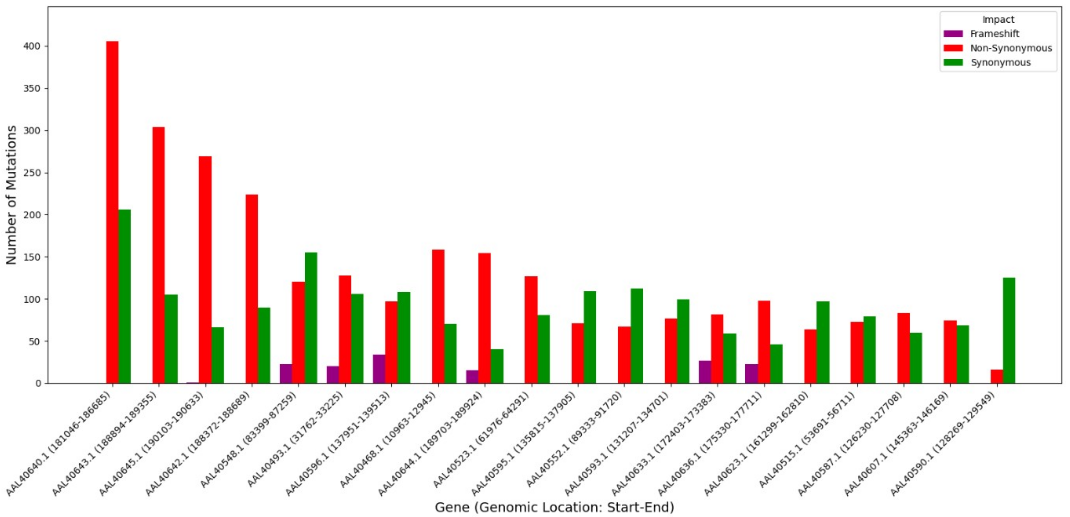
Genomic Analysis of Monkeypox Virus Variants
The Challenge: The re-emergence of Monkeypox (hmpxv) as a global threat has highlighted critical gaps in understanding its genetic evolution, especially within its African endemic regions. A lack of genomic surveillance hinders the ability to track transmission, predict virulence, and inform public health responses.
My Approach: I designed and executed a full bioinformatics pipeline to analyze 23 complete hmpxv genomes from six African countries. This involved data retrieval from GISAID, rigorous quality control in Python, multiple sequence alignment using MAFFT, and variant calling with FreeBayes. I then performed functional annotation to assess the impact of mutations and reconstructed the virus's evolutionary history using Maximum Likelihood phylogenetic analysis in MEGA X.
The Outcome: The analysis identified over 18,000 genetic variants, with 62.5% of coding mutations being non-synonymous, indicating strong potential for altered viral function. The phylogenetic analysis clearly delineated the major Clade I (Congo Basin) and Clade II (West African) lineages, revealing distinct, geographically-linked evolutionary paths. This study provides a crucial genomic baseline and a computational framework for monitoring emerging viral threats in real-time.
View Code on GitHub